


RESTseq - Population Genomics
RESTseq (Restriction fragment sequencing), an efficient genotyping by sequencing approach for Population Genomics on benchtop and other next generation sequencing platforms (e. g. Ion Torrent PGM).
The paper was published in PLoS ONE :
Efficient Benchtop Population Genomics with RESTriction Fragment SEQuencing
Abstract
We present RESTseq, an improved approach for a cost efficient, highly flexible and repeatable enrichment of DNA fragments from digested genomic DNA using Next Generation Sequencing platforms including small scale Personal Genome sequencers. Easy adjustments make it suitable for a wide range of studies requiring SNP detection or SNP genotyping from fine-scale linkage mapping to population genomics and population genetics also in non-model organisms. We demonstrate the validity of our approach by comparing two honeybee and several stingless bee samples.
Citation : Stolle E, Moritz RFA (2013) RESTseq Efficient Benchtop Population Genomics with RESTriction Fragment SEQuencing. PLoS ONE 8(5):e63960.
doi:10.1371/journal.pone.0063960
Media coverage / Awards
Article in GenomeWeb: German Team Develops Genotyping-by-Sequencing Approach for Desktop Machines (full text )
Finalist for the Ion Torrent Breakthrough Award 2013
further info
We recently developed and published a novel approach for genotyping-by-sequencing on benchtop next-generation sequencers for population genomic studies by efficiently generating sequencing libraries which are strongly reduced.
Our aim was, to have a flexible and robust tool for an efficient reduction of genomic libraries. Similar approaches (RAD, double-digest RAD, 2b-RAD, GBS, CRoPS etc.) also employ Restriction Endonucleases and subsequent steps for library preparation. However, there are still too many fragments to be sequenced, or (if rare cutting enzymes are used) the fragment distribution across the genome is more biased. Especially for small-scale benchtop machines this was not feasable. In addition, some applications do not require the generation of thousands of SNP markers, but might suffice with a couple of hundred.
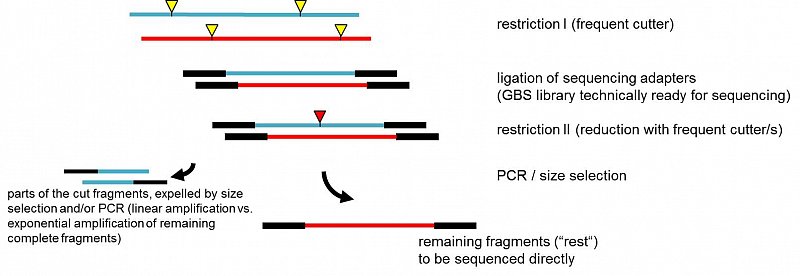
This lead us to use a very frequent cutting enzyme, in our experiments TaqI (TCGA), to generate the first library. Even with size selection, the fragments would be too numerous for a benchtop machine, although technically possible. But they also show a very dense and unbiased distribution across the genome.
The difference to previous approaches is, that we now digest the first library again with additional restriction enzyme(s) (of course chosen to not cut the (barcoded) adapter). This cuts a certain number of the TaqI-TaqI fragments, which subsequently will be expelled by size selection (the partial fragments are usually smaller than the left-over complete fragments) and/or in the PCR step: only complete fragments (TaqI-TaqI - same adapter on both sides) will be amplified exponentially, whereas any partial fragment (only on one side a TaqI adapter, if at all) will be amplified linearly. Thus, the complete remaining fragments, the "rest", will be highly overrepresented and are then ready to be sequenced.
The user has full control on how much he wants to reduced the initial (TaqI)library. The more frequent the second cutter is, the stronger the reduction. One can also combine several enzymes in the second step or enzymes which are specifically cutting a sequence of interest (lets say a very rare cutter which has also a site in a very frequent transposable or other repetitive element which is otherwise over-represented in the sequencing data). In our published test set-ups we digested the TaqI-library with MseI(TruI) (TTAA). This should be especially usefull in species whose genomes are AT-rich. AT-rich fragments, which might be problematic to be sequenced anyway, will be cleaved and will be expelled. In another set-up we combined MseI, MluCI, HaeIII, MspI and HinP1I in the second digestion (all 37°C, buffer NEB4) - thus fragments containing TTAA, AATT, GGCC, CCGG or GCGC were cleaved and expelled. This way the complexity of the library was reduced much more (similar as shown in fig.1 in the paper) and AT-/ GC-rich sequences were avoided to be sequenced. With this reduction, even barcoding and pooling of many samples work for the comparatively smaller capacity of benchtop platforms.
steps to prepare your library
Further example figures on Figshare


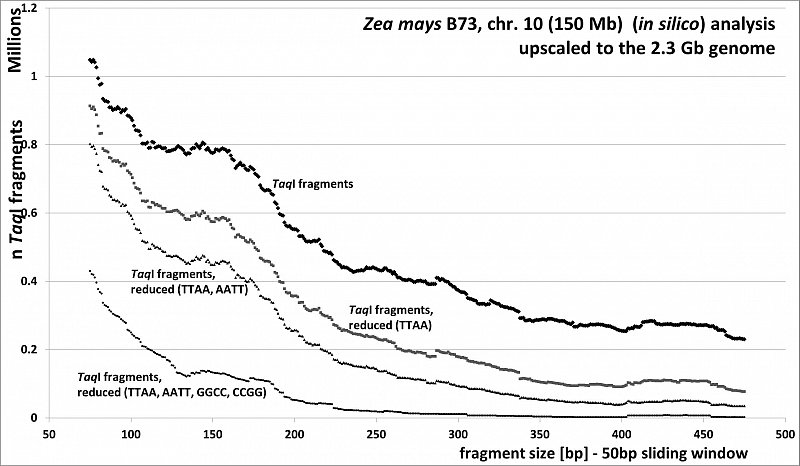
Zea mays (46.8% GC, ca 80% TE's), RESTseq example, fragment number in relation to selected fragment size and after reduction with different RE's, based on 150Mb chr 10, upscaled to the 2.3Gb genome
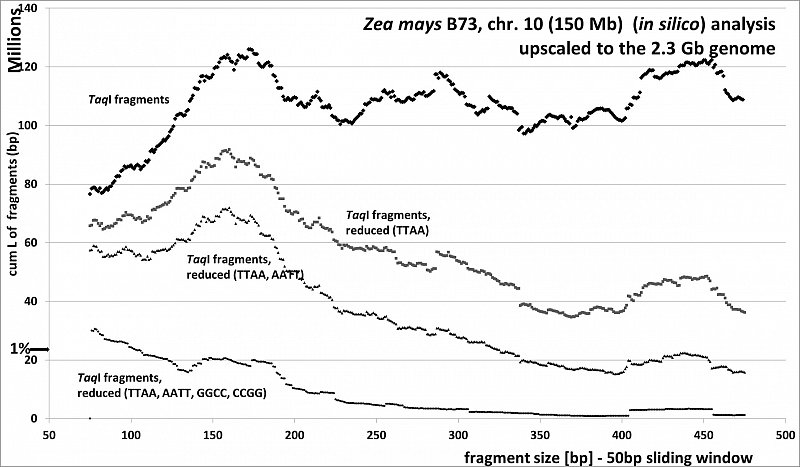
Zea mays (46.8% GC, ca 80% TE's), RESTseq example, cumulative length of remaining fragments in relation to selected fragment size and after reduction with different RE's, based on 150Mb chr 10, upscaled to the 2.3Gb genome
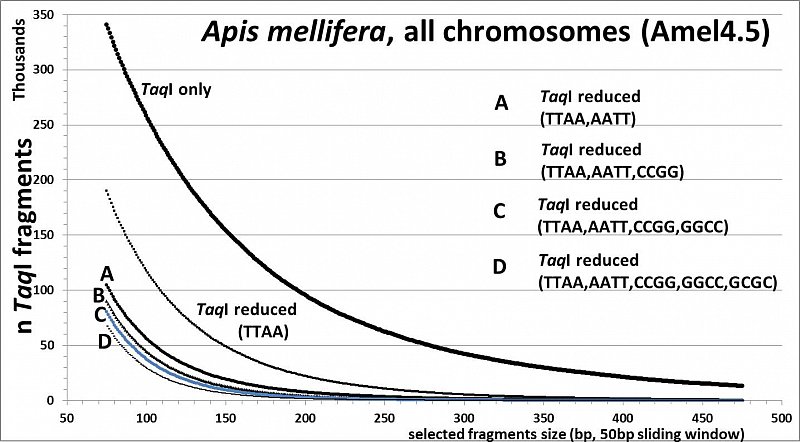
Apis mellifera (32.7% GC, <10% TE's), RESTseq example, fragment number in relation to selected fragment size and after reduction with different RE's, based on whole genome in silico analysis (Amel4.5, all 16 chr)
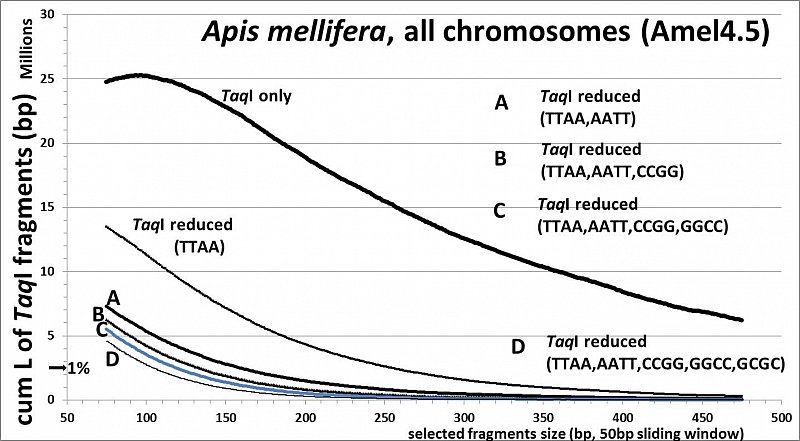
Apis mellifera (32.7% GC, <10% TE's), RESTseq example, cumulative length of remaining fragments in relation to selected fragment size and after reduction with different RE's, based on whole genome in silico analysis (Amel4.5, all 16 chr)
We will add more information here very soon.
Examples from a run of a RESTseq (TaqI, T^CGA) library prepared from a stingless bee female, reduced with MseI (T^TAA)
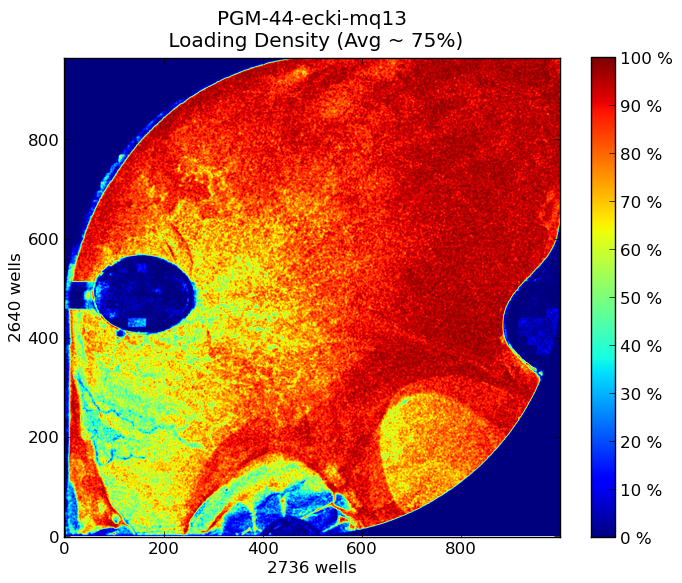
Ion Torrent - stingless bee RESTseq run on a 316 chip - bead density after loading the sample
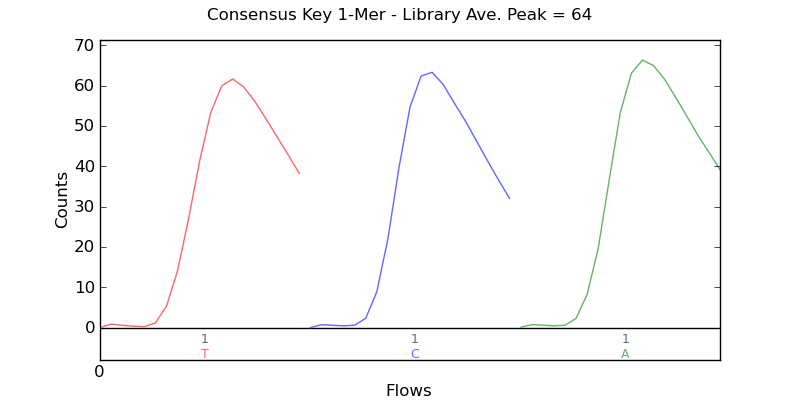
Ion Torrent - stingless bee RESTseq run on a 316 chip - key signal

Ion Torrent - stingless bee RESTseq run on a 316 chip - internal control fragment
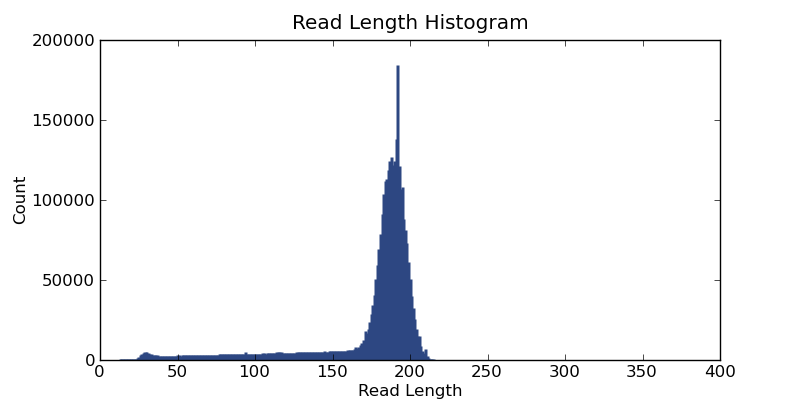
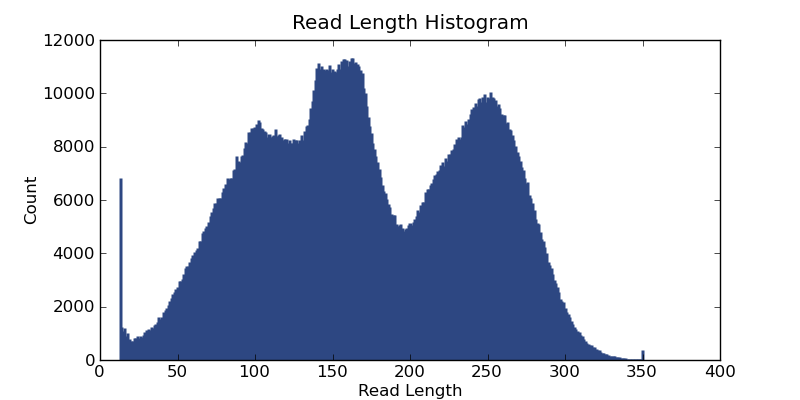
Ion Torrent - stingless bee RESTseq run on a 316 chip - read length distribution - the fragment library was size selected for about 160-205 bp
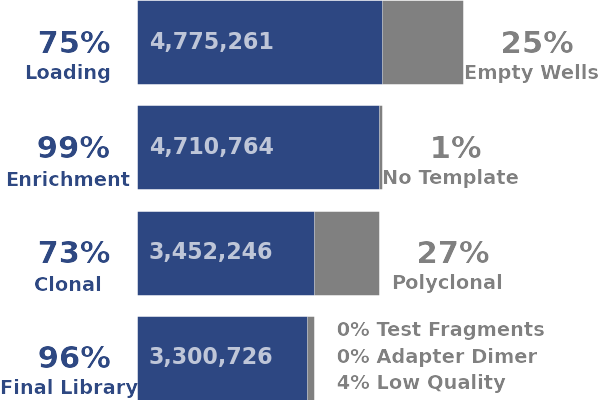
Ion Torrent - stingless bee RESTseq run on a 316 chip - summary stats for the sequencing run
Ion Torrent - stingless bee RESTseq run on a 316 chip - trimming of the barcode sequence, 97.96% of the reads started with the barcode.
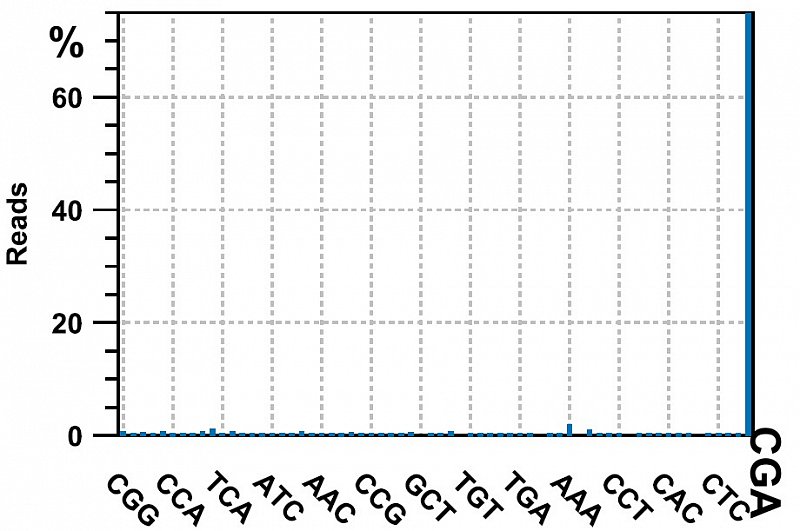
Ion Torrent - stingless bee RESTseq run on a 316 chip - check for the presence of the CGA triplet on both side of the reads (from the TaqI site: T^CGA). If it is present on both side, its an indicator that this fragment was completely sequenced. 75% of the reads contained CGA on both sides, the rest was discarded.
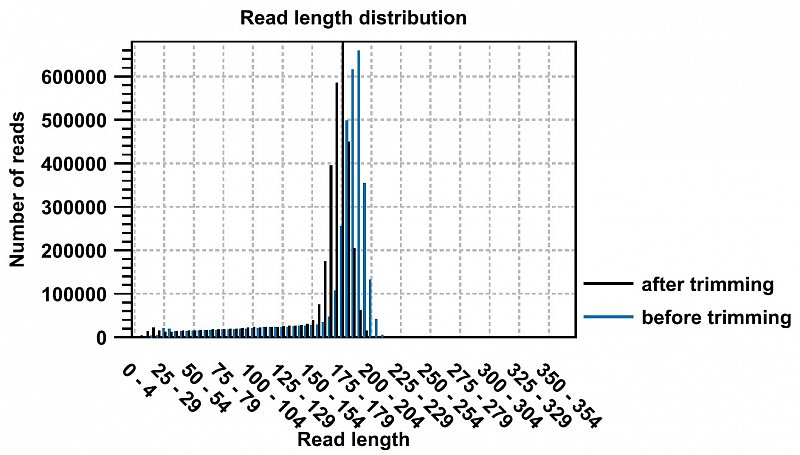
Ion Torrent - stingless bee RESTseq run on a 316 chip - removal of reads short/larger than the roughly targeted range of 160-210. reads smaller than 130 and larger than 250 were discarded - 99.65% were in the desired range.
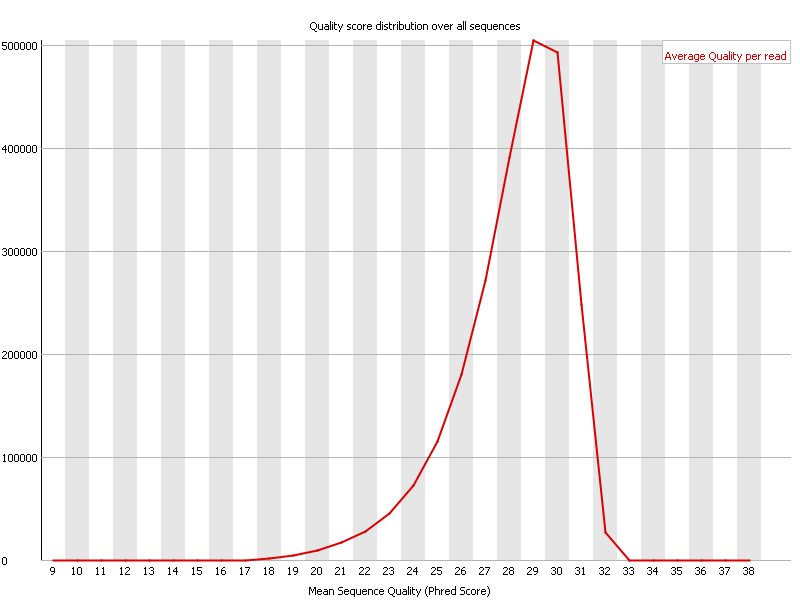
Ion Torrent - stingless bee RESTseq run on a 316 chip - FASTQC analysis on the final set of reads (trimmed barcode, CGA reads 130-250bp) - quality distribution
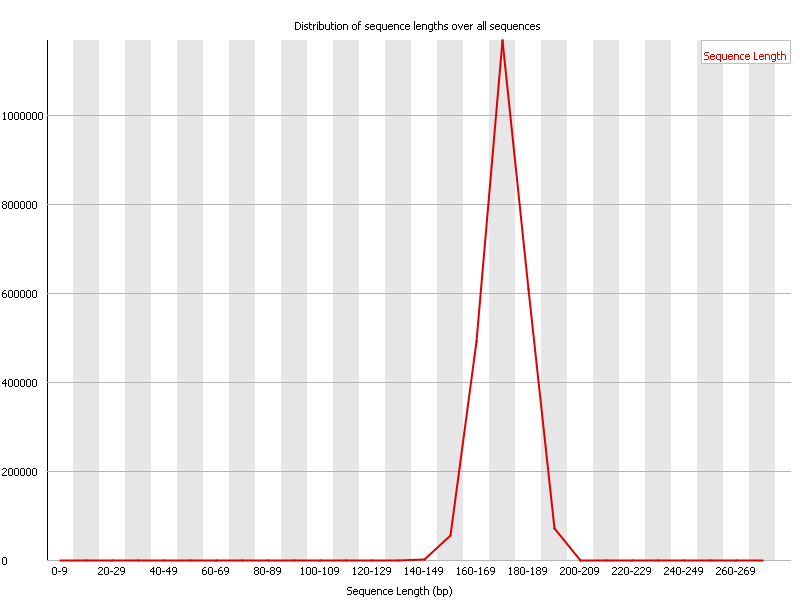
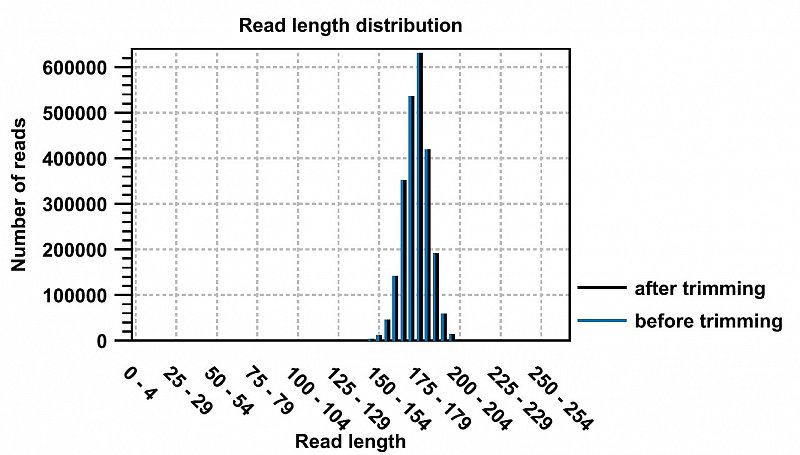
Ion Torrent - stingless bee RESTseq run on a 316 chip - FASTQC analysis on the final set of reads (trimmed barcode, CGA reads 130-250bp) - length distribution
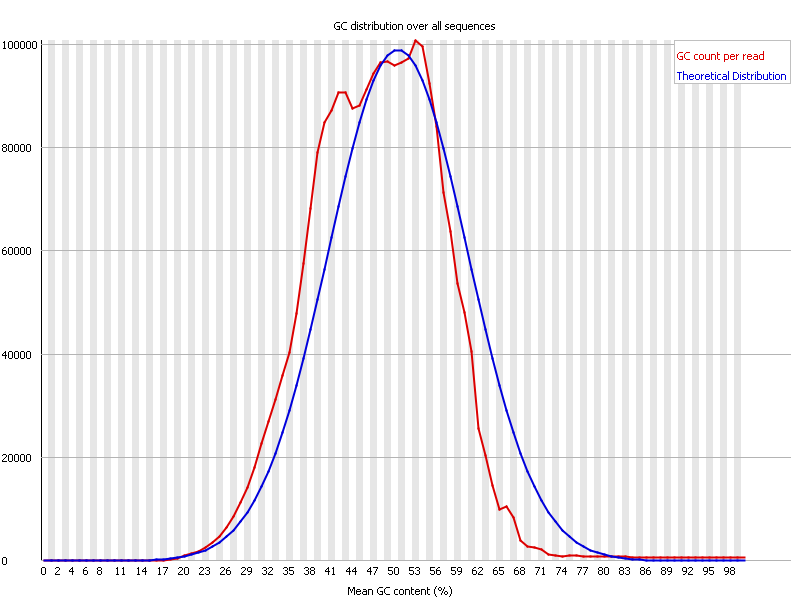
Ion Torrent - stingless bee RESTseq run on a 316 chip - FASTQC analysis on the final set of reads (trimmed barcode, CGA reads 130-250bp) - GC content
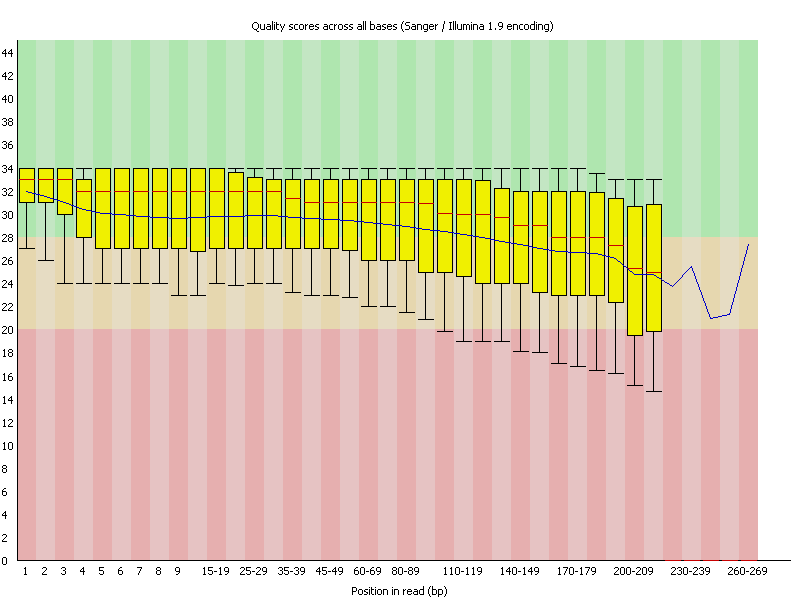
Ion Torrent - stingless bee RESTseq run on a 316 chip - FASTQC analysis on the final set of reads (trimmed barcode, CGA reads 130-250bp) - per base quality
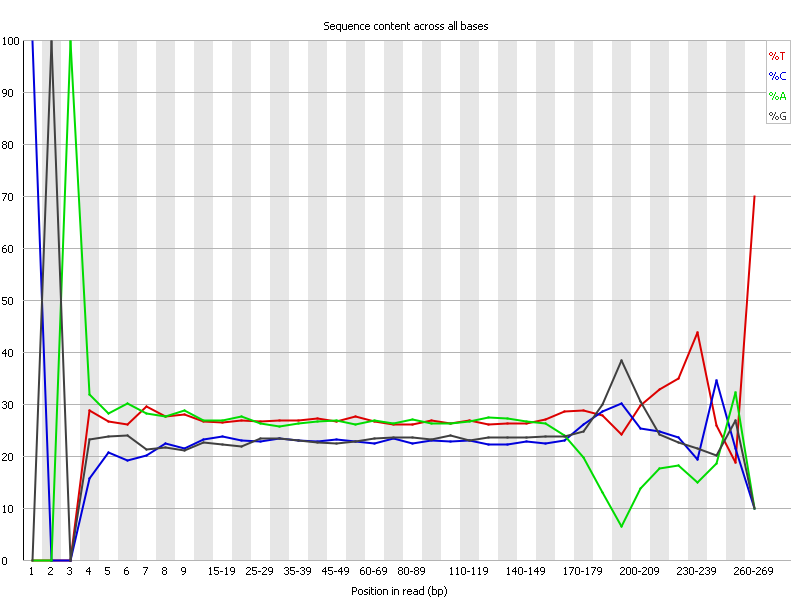
Ion Torrent - stingless bee RESTseq run on a 316 chip - FASTQC analysis on the final set of reads (trimmed barcode, CGA reads 130-250bp) - per base nucleotide content - note the CGA at the beginning and end (variable read length, thus variable position of the CGA there) of each read
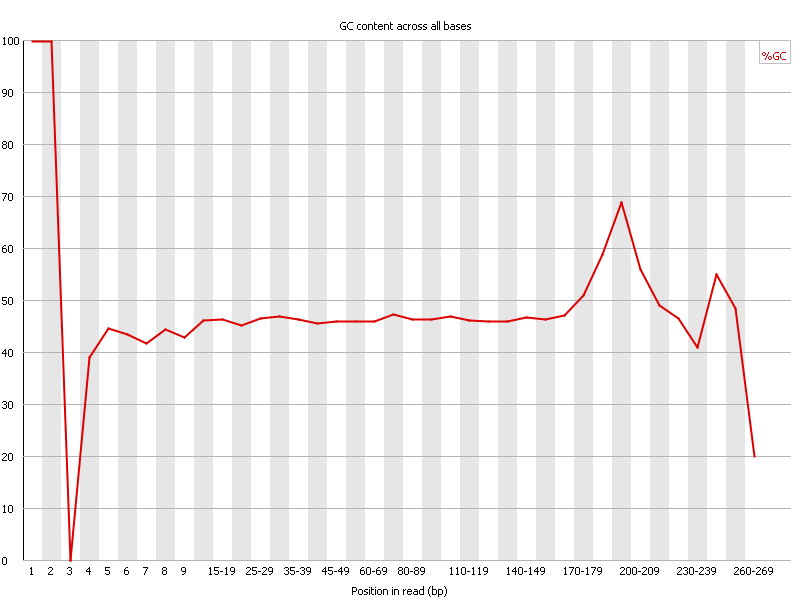
Ion Torrent - stingless bee RESTseq run on a 316 chip - FASTQC analysis on the final set of reads (trimmed barcode, CGA reads 130-250bp) - per base GC content - note the CGA at the beginning and end (variable read length, thus variable position of the CGA there) of each read.
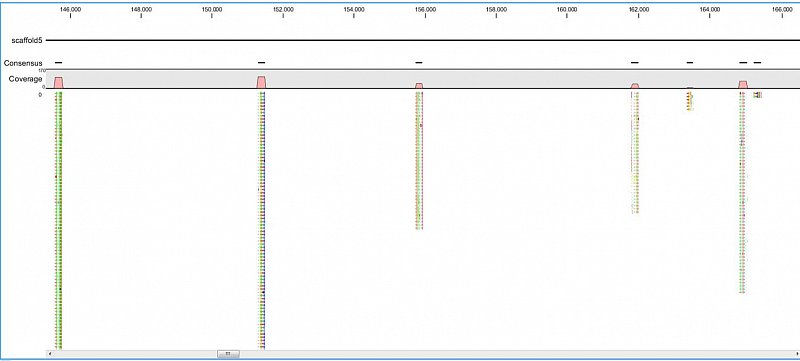
Ion Torrent - stingless bee RESTseq run on a 316 chip - mapping on a reference genome of a related species.
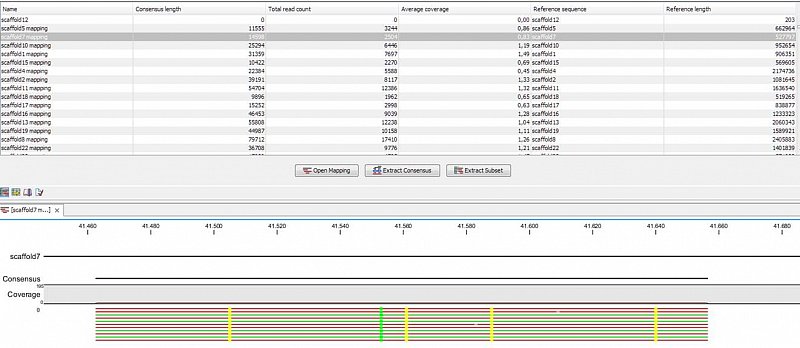
Ion Torrent - stingless bee RESTseq run on a 316 chip - mapping on a reference genome of a related species.
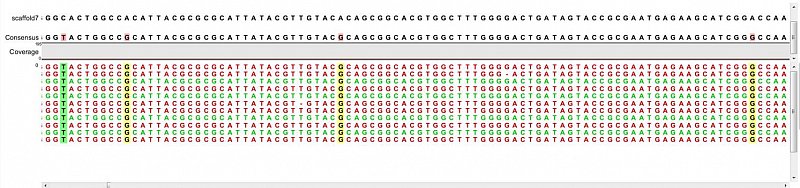
Ion Torrent - stingless bee RESTseq run on a 316 chip - mapping on a reference genome of a related species.
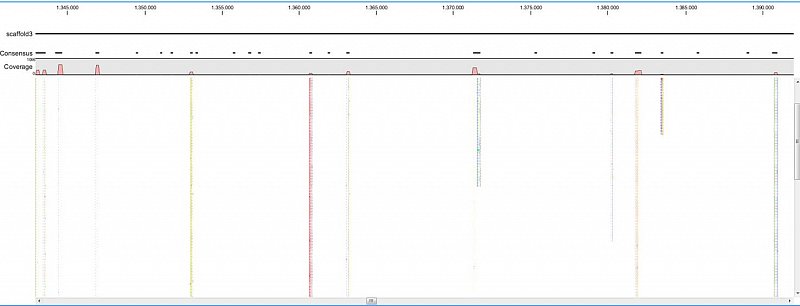
Ion Torrent - stingless bee RESTseq run on a 316 chip - mapping on a reference genome of a related species. Shown in the picture is a ~50kb region with a dozen of fragments sequenced.

Ion Torrent - stingless bee RESTseq run on a 316 chip - mapping on a reference genome of a related species. This example shows the reads from a certain fragment containing homozygous and one heterozygous SNP.
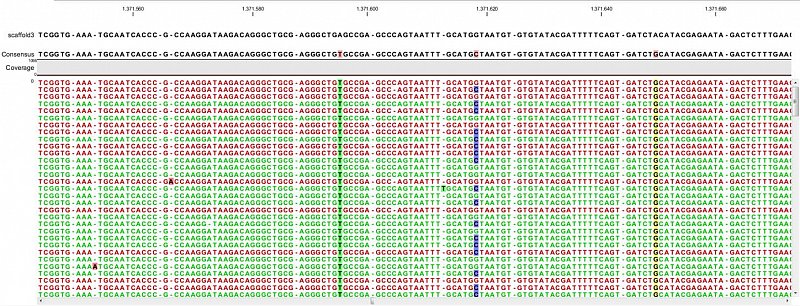
Ion Torrent - stingless bee RESTseq run on a 316 chip - mapping on a reference genome of a related species. G/C SNP (heterozygous diploid female)
Ion Torrent - different stingless bee RESTseq run on a 316 chip. This is an example of a bad run. The library was overamplified and this resulted in a bad quality (abrupted, <130bp) of many reads (targeted fragment size 130-180) and concatemers (larger than 200bp). Especially the concatemers cannot be used for analysis, yet they occupy a large fraction of the chip's capacity. Options to circumvent this problem is a reduction of the cyclenumer used for amplification of the library (<8-10) (e.g. by starting with more material), or to do the (or an additional) sizeselection step after the PCR (works well in our experiments).
further reading
RAD (Baird et al. 2008)
2b-RAD (Wang et al. 2012
Double Digest RADseq (Peterson et al. 2012)
GBS (Elshire et al. 2011)
RRL (Tassel et al. 2008)
modified RRL (Luca et al. 2011)
CRoPS (Orsouw et al. 2007)
Multiplexed shotgun genotyping (Andolfatto et al. 2011)
Paired-end RAD-seq (Willing et al. 2011)
Local Assemblies of Paired-End Reduced Representation Libraries (Deschamps et al. 2012)
analysis tools
Currently, a specific software solution for the clustering and analysis of RESTseq fragments is being developed
other useful tools:
RADseq tools
STACKS software
STACKS paper (Catchen et al. 2011)
STACKS paper (Catchen et al. 2013)
TASSEL pipeline
GBS info
CLC genomics workbench
Rainbow, paper - Clustering/assembly tool for RADseq reads (Chong et al. 2012)
Rainbow - Clustering/assembly tool for RADseq reads
CORTEX tool for assembly and variation analysis
CORTEX paper: Iqbal et al. 2011: De novo assembly and genotyping of variants using colored de Bruijn graphs
SEQanswers forum
Vmatch - Willing et al. 2011, RApiD pipeline, tool 1(clustering)
LOCAS - Willing et al. 2011, RApiD pipeline, tool 2
this page and the containing images were created by Eckart Stolle
Besucherzähler